Що означає діагноз фенілкетонурія і небезпечна ця хвороба для здоров’я дитини?
Дізнавшись, що це за захворювання – фенілкетонурія, діагностування якого проводиться ще в період новонародженості, при його виявленні потрібно негайно приступити до початку лікування. Раннє виявлення і терапія дозволяють досягти сприятливих результатів.
Історія
З’ясувалося, що за розвиток хвороби відповідає лише один ген, який отримав найменування РАН (ген фенілаланінгідроксилази).
Завдяки цій знахідці вченим і лікарям по всьому світу вдалося більш детально виділити і описати як саме захворювання, так і його симптоми і форми. Більш того, були знайдені і розроблені абсолютно нові, високотехнологічні і сучасні способи лікування, такі, як генотерапія, яка на сьогодні є зразком ефективної боротьби з генетичними патологіями людини.

патогенез
В результаті викликаного генним дефектом порушення обміну фенілаланіну і його подальшого перетворення в тирозин в організмі накопичуються токсичні похідні цієї амінокислоти (фенилпировиноградная, фенілмолочная і фенилуксусная кислоти), в нормі зустрічаються в мінімальній кількості. Утворюються також не зустрічаються в нормі ортофенілацетат і фенілетиламін, що порушують ліпідний обмін в головному мозку, що служить причиною прогресуючого зниження інтелекту.
Факторами, що впливають на розвиток патології, є:
- порушення амінокислотного обміну;
- порушення мієлінізації;
- порушення синтезу протеоліпідним білків,
- знижений рівень нейротрансмітерів (адреналіну, серотоніну та ін.).
Механізм розвитку і причини захворювання
Причина виникнення цього захворювання пов’язана з тим, що в печінці людини не виробляється особливий фермент – фенілаланін-4-гідроксилази. Він відповідає за перетворення фенілаланіну в тирозин. Останній входить до складу пігменту меланіну, ферментів, гормонів і необхідний для нормальної роботи організму.
При ФКУ фенілаланін, в результаті побічних шляхів обміну, перетворюється в речовини, яких не повинно бути в організмі: фенілпіровиноградну і фенілмолочная кислоти, фенілетиламін і ортофенілацетат. Ці сполуки накопичуються в крові і надають комплексну дію:
- порушують процеси жирового обміну в мозку;
- надають токсичну дію, отруюючи мозок;
- викликають дефіцит нейромедіаторів, які передають нервовий імпульс між клітинами нервової системи.
Це викликає значне і необоротне зниження інтелекту. У дитини швидко розвивається розумова відсталість – олігофренія.
Захворювання успадковується тільки в тому випадку, якщо обидва батьки передали дитині схильність до хвороби, і тому зустрічається досить рідко. У двох відсотків людей є змінений ген, який відповідає за розвиток хвороби. При цьому людина залишається повністю здоровим. Але коли чоловік і жінка, носії гена, що мутує, одружуються і вирішують завести дітей, то ймовірність того, що малюки будуть страждати від фенілкетонурії, становить 25%. А можливість того, що діти будуть носіями патологічного гена ФКУ, але самі залишаться практично здоровими, становить 50%. [Adsen]
Опис хвороби, код за МКХ-10
Фенілкетонурія, або хвороба Фелінга, – це вроджена патологія, яка має генетичну природу.
Захворювання передбачає порушення гідроксилювання фенілаланіну, а також накопичення амінокислоти з продуктами її обміну в тканинах і рідинах організму з подальшим серйозним ураженням центральної нервової системи.
Захворювання вперше було описано І. Фелінга в 1934 р Воно досить рідкісне, зустрічається в одному випадку на 10 000 немовлят .
Хвороба може призвести до серйозних порушень у дитини. Бажано виявити його до того, як проявляться симптоми.
На даний момент це дуже важливе завдання для педіатрів, неонатологів і генетиків.
Код хвороби по МКБ-10 – E70. Порушення обміну ароматичних амінокислот.
Про те, що таке фенілкетонурія, розповість відео:
симптоми фенілкетонурії
Фенілкетонурія (див. Фото) проявляється на першому році життя. Основними симптомами в цьому віці є:
- млявість дитини;
- відрижки;
- порушення м’язового тонусу (частіше м’язова гіпотонія);
- судоми;
- відсутність інтересу до навколишнього;
- іноді підвищена дратівливість;
- занепокоєння;
- ознаки алергічного дерматиту;
- з’являється характерний «мишачий» запах сечі.
При фенілкетонурії характерні наступні фенотипічні особливості: гипопигментация шкіри, волосся, райдужної оболонки очей. У деяких хворих одним із проявів патології може бути склеродермія.
У більш пізньому віці для хворих на фенілкетонурію характерна затримка психомовного розвитку, нерідко відзначається мікроцефалія. Епілептичні напади зустрічаються майже у половини хворих на фенілкетонурію та в деяких випадках можуть служити першою ознакою хвороби. [Adsense2]
діагностика
Важливим, як ми вже зазначили, є раннє діагностування захворювання, що дозволить уникнути його розвитку і привести до ряду незворотних і важких наслідків. З цієї причини в пологових будинках до 4-5 днях життя (для новонароджених доношених) береться для аналізу кров. У недоношених дітей на предмет фенілкетонурії (ФКУ) кров береться на 7 день.
Процедура передбачає взяття капілярної крові через годину з моменту годування, нею зокрема просочується спеціальний бланк. Концентрація, яка вказує на позначку понад 2,2% фенілаланіну в крові малюка, вимагає направлення його з батьками для огляду в медико-генетичний центр. Там же проводиться дообстеження і, власне, уточнення діагнозу.
Як лікувати фенілкетонурію
Єдиним дієвим методом лікування фенілкетонурії вважається організована з перших днів життя спеціально розроблена дієта, принцип якої полягає в обмеженні міститься в продуктах харчування фенілаланіну, для чого виключаються такі продукти харчування як:
- крупи,
- бобові,
- яйця,
- сир,
- хлібобулочні вироби,
- горіхи,
- шоколад,
- риба, м’ясо та ін.
Лікувальний раціон хворих на фенілкетонурію складається зі спеціалізованих продуктів як зарубіжного, так і вітчизняного виробництва. Дітям першого року життя показані продукти, за своїм складом наближені до грудного молока, це такі суміші як «Лофенілак» і «Афенілак». Для дітей трохи старше розроблені такі суміші як «Тетрафен», «Максамум-ХР», «Феніл-Фрі». Тим, хто страждає на фенілкетонурію вагітним жінкам і дітям старшого віку (після шести років) показаний прийом суміші «Максамум-ХР». Крім спеціалізованих лікувальних продуктів в раціон хворого включають соки, фрукти і овочі.
Своєчасно розпочата дієтотерапія найчастіше дозволяє уникнути розвитку характерних клінічних проявів класичної фенілкетонурії. Лікування в обов’язковому порядку проводиться до статевого дозрівання, а іноді й довше. Внаслідок того, що хвора на фенілкетонурію жінка виносити здоровий плід не здатна, проводиться розпочате ще перед зачаттям і триваюче до самих пологів спеціальне лікування, спрямоване на виключення ураження плода фенилаланином від хворої матері.
Які перебувають на лікуванні діти повинні перебувати під невсипущим контролем психоневролога і дільничного педіатра. На початку лікування фенілкетонурії контроль вмісту фенілаланіну проводять щотижня, при нормалізації показників переходять на 1 раз на місяць протягом першого року життя, і 1 раз в два місяці у дітей старше року.
Крім дієтотерапії, дітям з фенілкетонурією лікарі можуть робити такі призначення:
- мінеральні сполуки;
- ноотропні засоби;
- вітаміни групи В;
- антиконвульсанти.
У комплексній терапії повинні бути присутніми лікувальна фізкультура, голкорефлексотерапія і масаж.
Зверніть увагу: при атипової формі фенілкетонурії, яка не піддається корекції дієтотерапією, лікарі призначають гепатопротектори, протисудомні засоби. Таке лікування допоможе полегшити стан дитини.
лікування
При своєчасно поставленому діагнозі лікування фенілкетонурії полягає в спеціалізованій дієті, яка обмежує надходження їжі, що містить фенілаланін. Так як всі природні джерела білка містять близько 4% фенілаланіну, вони замінюються містять інші амінокислоти синтетичними продуктами. Найбільш ефективна дієта, призначена до 8-го тижня життя.
Немовлятам призначаються повністю очищені від лактози суміші на основі гідролізату молочного білка. Материнське молоко допустимо в обмеженій кількості.
Хоча раніше лікарі рекомендували дотримуватися дієти тільки до закінчення розвитку головного мозку (20 років), підвищення рівня фенілаланіну після припинення дієти у багатьох людей викликає психіатричні проблеми. Фенілкетонурія у дорослих проявляється недостатністю мотивації, безсонням і деконцентрації, імпульсивністю і т.д., тому дієту рекомендується дотримуватися довічно.
Дієтичні обмеження, введені після 2-х років, здатні тільки знизити вираженість симптомів.
Для лікування застосовуються також препарати:
- які стосуються ноотропной групі (ноотропіл та ін.). Вони стимулюють розумову діяльність і активізують когнітивні функції;
- містять вітаміни, амінокислоти і білки ( «Афенілак» і т.д.).
Атипові форми фенілкетонурії не піддаються дієтотерапії і вимагають додаткового медикаментозного лікування, яке включає:
- Дігідробіоптерін, компенсуючий недолік фолієвої кислоти;
- Леводол, діючий на ЦНС.
Пацієнтам, які потребують додаткової корекції нейромедіаторів, призначають «Мадопар» або «НАКом».
Дорослим призначаються рослинні препарати, що містять фенілаланінгідроксилази.
Фенілкетонурія і материнство
Для вагітних жінок, хворих на ФКУ, дуже важливо утримувати низький рівень фенілаланіну до і під час всієї вагітності, для того, щоб дитина була здоровою. І хоча, плід, що розвивається може бути тільки носієм гена ФКУ, проте внутрішньоутробна середовище може мати дуже високий рівень фенілаланіну, який має здатність проникати через плаценту. Як наслідок, у дитини може розвиватися вроджений порок серця, можлива затримка розвитку, мікроцефалія і розумова відсталість. Як правило, у хворих на фенілкетонурію жінок ніяких ускладнень під час вагітності не виникає.
У більшості країн, жінкам з ФКУ, які планують заводити дітей, рекомендується знизити рівень фенілаланіну (як правило до 2-6 мкмоль / л), ще до вагітності і контролювати його протягом усього періоду виношування дитини. Це досягається шляхом проведення регулярних аналізів крові і дотримання суворої дієти, і постійним наглядом лікаря-дієтолога. У багатьох випадках, як тільки печінку плода починає нормально виробляти PAH, рівень фенілаланіну в крові матері падає, відповідно «необхідно» збільшити його, для утримання безпечного рівня – 2-6 мкмоль / л.
Саме тому, щоденна кількість спожитого матір’ю фенілаланіну може подвоїтися або навіть потроїтися до кінця вагітності. Якщо ж рівень фенілаланіну в крові матері нижче 2 мкмоль / л, то іноді, у жінок можуть виникати різні ускладнення, пов’язані з дефіцитом цієї амінокислоти, такі як головний біль, нудота, випадання волосся і загальне нездужання. Якщо низький рівень фенілаланіну у хворих ФКУ підтримується протягом всієї вагітності, то ризик народити хвору дитину не вище, ніж у тих жінок, які не хворі ФКУ. [Adsense3]
види
Фенілаланін під впливом ферментів в організмі перетворюється в тирозин – амінокислоту, яка виводиться з організму. Залежно від дефекту гена, що блокує певний фермент, виділяють:
- Фенілкетонурію I-го типу (класичну або важку). Викликається генною мутацією, що порушує вироблення печінкового ферменту фенілаланінгідроксилази і перетворення фенілаланіну в тирозин.
- Фенілкетонурію II-го типу (атипову). Характеризується генним дефектом, що викликає недолік дігідробіоптерін-редуктази. Завдяки цьому фактору порушується відновлення активності органічної сполуки, необхідного для перетворення фенілаланіну. Також спостерігається знижений вміст у спинномозковій рідині і в сироватці крові вітаміну В9, необхідного для утилізації амінокислот.
- Фенілкетонурію III-го типу (атипову). Провокується нестачею каталізатора, необхідного для синтезу тетрагідробіоптеріна (потрібен для перетворення фенілаланіну в тирозин).
- Прімаптерінурію – атипову форму, яка спостерігається при легкій формі гіперфенілаланінемії. Ферментний дефект цього виду ФКУ в даний час не з’ясований, але дана форма захворювання характеризується значною кількістю прімаптеріна і його похідних в сечі, а кількість нейромедіаторних метаболітів в спинномозковій рідині не відхиляється від норми.
Виділяється також материнська фенілкетонурія, що спостерігається у потомства жінок, хворих на ФКУ і які не дотримуються спеціалізовану дієту. Патогенез даної форми захворювання до кінця не вивчений, але відзначається, що без постійного контролю за рівнем фенілаланіну у новонароджених виявляється ряд патологічних змін:
- недостатня вага мозку;
- збільшені в розмірах шлуночки головного мозку (вентрікуломегалія);
- гіпоплазія (недорозвинення) білої речовини і затримка мієлінізації.
Фенілкетонурія даного типу викликає хронічну інтоксикацію плоду і призводить до розумової відсталості дитини.
Наслідки і прогноз життя
Вплив зайвої кількості фенілаланіну на нервову систему дитини призводять до стійких психологічних порушень. Вже до 4 років, без належного лікування, хворі на фенілкетонурію діти зараховуються до недоумкуватим і фізично недорозвиненим членам суспільства. Вони поповнюють ряди інвалідів дитинства та фарби життя меркнуть для них.
Чи не іскриться щастям і життя батьків хворої дитини. Малюк вимагає постійного догляду, і при обмежених фінансових коштах це виливається в загальне погіршення добробуту сім’ї. Біль, яку відчувають мамою і татом від неможливості змінити існування дитини в кращу сторону пригнічує і тисне, але впадати у відчай не можна. Допоможіть собі, допоможіть дитині пройти ці випробування з меншими втратами в любові і милосердя.
Наука поспішає, вона робить семимильні кроки в напрямку виключення захворювання з рангу важких. Величезне значення має діагностика фенілкетонурії в утробі матері, але поки такого методу не винайдено. «Поки» не означає «ніколи», будемо чекати і вірити
спадкування
Так як фенілкетонурія успадковується як рецесивна ознака, то для прояву його у дитини необхідно, щоб у обох батьків був дефектний ген. Саме тому родинні шлюби в багатьох країнах заборонені.
Якщо розглядати випадок народження дітей в звичайній родині, то у носіїв такої мутації вони можуть бути:
- 25% ймовірності, що дитина народиться хворою.
- У 50% випадків маля здорове, але є носієм дефектного гена.
- Четверта частина потомства буде абсолютно нормальною.
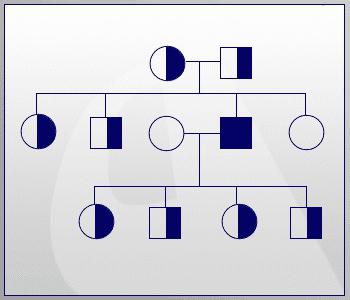
Ця схема не дає повної картини народжуваності хворих дітей. Вона тільки відображає ймовірність, тому у кожної сімейної пари відсоток дефектних генів може бути свій, і передбачити результат, на жаль, неможливо. Зараз існують консультації, в яких фахівці-генетики допомагають парам спрогнозувати народження у них хворої дитини, розповідаючи при цьому, як фенілкетонурія успадковується.