Ознаки синдрому Ретта, лікування, прогноз і діагностика
Синдром Ретта у дітей – що це таке і в чому небезпека?

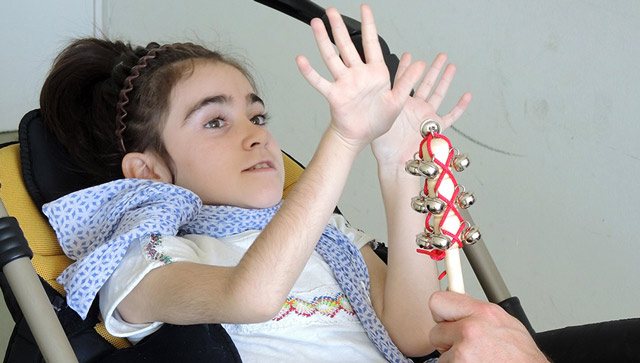
Фото 1. Дівчинка з синдромом Ретта
Синдром Ретта – рідко зустрічається генетичне захворювання, одним з головних ознак є ураження центральної нервової системи, яке тягне за собою втрату розумових і фізичних навичок, які людина придбала протягом свого життя. Код МКХ-10 F84.2.
Так як це мутація конкретно Х-хромосоми, то це захворювання вважається летальним для хлопчиків, так як у них одна Х-хромосома. У дівчаток їх дві, тому загроза житті не настільки велика. Синдром Ретта тягне за собою розумову недорозвиненість і відставання у фізичному розвитку.
Вперше описав цю хворобу і виділив основні симптоми Андреас Ретт в 1960-1970 роки. Але особливо на цю хромосомну патологію звернули увагу група вчених, які під керівництвом Бенгта Хагберга зробили публікацію. Саме ці вчені і лікарі описали симптоми деменції (розвиток недоумства), ознаки аутизму, порушення в м’язовій системі і в русі рук у дівчаток. Дослідження проводили відразу в трьох країнах Європи. Саме наукова робота і спостереження цих фахівців допомогли медицині виділити синдром Ретта, як окреме захворювання.
історичні відомості
У 1954 році названий вище австрійський лікар Андреас Ретт обстежив двох дівчаток, в результаті чого зазначив у них крім затримки психічного розвитку деякі стереотипні рухи, що нагадують «миття рук» або в вигляді «стискання рук». Він відшукав кілька подібних випадків в своїх записах, що наштовхнуло його на думку про унікальність даного захворювання. Доктор Ретт, знявши своїх пацієнток на відеоплівку, відправився в пошуках дітей з подібними симптомами по всій Європі.
У 1966 році він опублікував свої дослідження в Австрії в парі німецьких журналів. Однак всесвітньої розголосу вони не отримали, навіть після публікації в 1977 році англійською мовою. Лише після публікації шведського дослідника Б. Хагберга і його колег в 1983 році захворювання було названо на честь свого першовідкривача «синдромом Ретта» і виділено в окрему нозологічну одиницю.
причини
Під цією хворобою розуміють зміна (мутацію) гена MECP2 на Х-хромосомі, яка визначає стать людини, разом з У-хромосомою. Жіночої статі характерно дві Х-хромосоми, а представники чоловічої статі одну Х і одну У хромосоми. Хвороба вражає в основному дівчаток, тому що у них є друга копія гена MECP2, яка функціонує, як годиться, і дитина виживає.
Причини походження синдрому Ретта не до кінця вивчені, незважаючи на те, що вчені довели пряме нахил спадковості до даної хвороби. Тому є кілька припущень, що пояснюють розвиток синдрому Ретта, але і вони до кінця не є точними і вірними:
- мутації на генному рівні, спровоковані високим кількістю родинних зв’язків в поколінні (2,7%, при статистикою в допустимому форматі 0,51%). З’являються вони ще в самому зародку, коли у плода формуються свої внутрішні органи;
- хромосомні невідповідності. Фахівці дотримуються думки, що проблема мешкає в Х-хромосомі, проте з’ясувати яку саме ділянку хромосоми патологічний не виходить;
- метаболічні порушення і дисфункція мітохондрій. У клінічних випадках фахівці помітили, що у хворих з синдромом Ретта спостерігаються зміни в картині крові. Підвищується рівень міоцитів і лімфоцитів, також перевищують норму молочна і піровиноградна кислота.
На захворювання впливають всі фактори, можливо вчені знайдуть більш детальні причини синдрому Ретта. Але патогенез у дитини залишається незмінним – припинення розвитку головного мозку, це впливає на зростання, розвиток внутрішніх органів, міопатію і розвиток супутніх хвороб.
Основні причини
Як тільки патологію винесли в окреме захворювання, фахівці почали висувати різні теорії причин її розвитку. Спочатку припустили, що недуга відрізняється генетичною природою, тобто у всьому винна мутація генів. Відхилення такого характеру пояснюються присутністю великої кількості кровних зв’язків в родоводу людини. З іншого боку, висловлювалися припущення про хромосомних аномаліях в якості основної причини захворювання. Тут мова йде про наявність ламкого ділянки в короткому плечі Х-хромосоми. Вчені припускають, що саме ця зона відповідальна за формування патології. Проведені згодом дослідження на цю тему довели, що у пацієнтів з таким діагнозом насправді є деякі порушення в хромосомах. Чи справді цей фактор є головною причиною психічних відхилень, до сих пір залишається невідомо. Єдине, що вдалося встановити точно, це вік хворих. На думку лікарів, первинні порушення в роботі мозку виникають з самого народження дитини, а вже до четвертого його році життя розвиток повністю припиняється. Більш того, такі діти не можуть повноцінно розвиватися і в фізичному сенсі.

Симптоми у дітей
Симптоми клінічно не визначаються в перші місяці життя дитини, але в окремих випадках спостерігається м’язова слабкість, початок повзання, сидіння і ходьби трохи пізніше, ніж у здорових дітей такого ж віку. Ознаки такі:
- Мікроцефалія. Дитина народжується з нормальною окружністю голови, але потім вона починає відставати в рості.
- Рухові порушення. У період від двох місяців до п’яти років у дитини помічають нездатність утримати пляшечку або іграшки. Також з’являються специфічні руху – «миття рук». Саме ця ознака є патогномонічним для синдрому Ретта. Спостерігається ляскання в долоні, стискання, постукування. Дитина кусає руки і стукає ними по тілу.
- Розумова відсталість. Всі ці зміни можна простежити за допомогою психологічних тестів. Всі ті навички мови і адаптації, які дитина придбав, зникають протягом декількох місяців.
- Порушення координації і їх спрямованість (атаксія і апраксія). Характерний тремор, втрата рівноваги, хитка хода і уривчасті руху.
- Розлади дихання. Апное (1-2 хвилини аж до непритомності) і гіпервентиляції. Під час сну дані симптоми не виявляються.
- Судомні напади. Тоніко-клінічні судоми та епілептичні припадки.
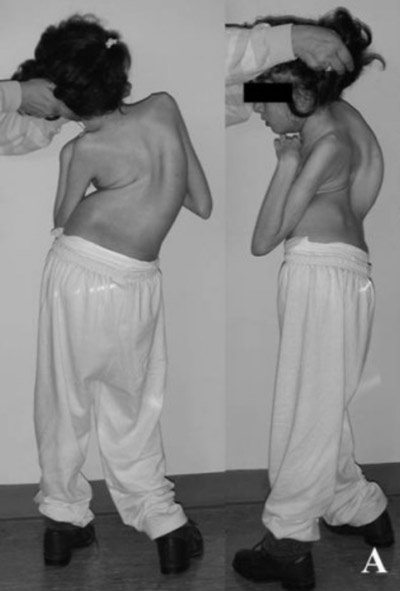
- Сколіоз. Розвивається це ускладнення через гіпотонії м’язів спини.
Всі перераховані вище симптоми синдроми Ретта поглиблюються з дорослішанням дитини. А якщо не застосовувати симптоматичне лікування, то стан буде погіршуватися з неймовірною швидкістю.
симптоми
Перші клінічні ознаки патології виникають до кінця першого року життя. До року дитина з синдромом Ретта зростає і розвивається звичайними темпами.

Перші ознаки недуги:
- гіпотермія,
- гіпергідроз,
- Блідість шкіри,
- Труднощі при перевертанні з живота на спину,
- Проблеми з повзання і сидінням.
Хворі діти відстають від однолітків у зростанні, мають непропорційні руки і ноги по відношенню до тулуба, пізно починають ходити, часто відригують, швидко і поверхнево дихають, страждають епізодами раптової зупинки дихання і судорожними припадками, мають затримку моторики й мови.
У хворих дітей виникають специфічні руху рук, пропадають навички утримування предметів, з’являються монотонні рухи – перебирання пальців або хлопки на рівні грудей, стискання кулачків, сплетіння пальців, піднесення рук до обличчя, засування їх в рот, покусування рук або удари ними по різних частинах тіла . Рухи рук повністю не зникають, вони стають незвичайними. Ментальне розвиток характеризується розумовою відсталістю і відсутністю пізнавальної діяльності, швидкою втратою набутих навичок. У хворих розвивається мікроцефалія, з’являються судомні напади, формується сколіоз, обумовлений дистонією м’язів спини. Непропорційність голови по відношенню до інших частин тіла стає дуже помітною ближче до року.
Хворі діти асоціальні. Вони не реагують на будь-які зовнішні подразники і схожі своєю поведінкою на хворих на аутизм. Гіперзбудливість, нападоподібний плач, абсолютна безпорадність – характерні ознаки патології. Діти з синдромом Ретта можуть довго розгойдуватися з боку в бік і переступав з ноги на ногу. У деяких відзначається високою больовий поріг. Вони проявляють агресію по відношенню до себе і оточуючих: б’ють близьких і себе, кусають нігті, дряпаються.
До чотирирічного віку зростання мозку у хворих дітей зупиняється повністю. Це призводить до дисфункції вегетативної нервової системи. У дитини сповільнюється зростання скелета, атрофуються м’язи, перестають нормально працювати внутрішні органи.
Епіпріпадкі, безсоння і деменція з віком змінюються паркінсонізмом, стійкими порушеннями роботи серця і судин, втратою волосся. У хворих часто спостерігається дісморфізм особи, що надає їм особливий зовнішній вигляд. Жінки з синдромом Ретта фертильності, тобто здатні до зачаття дитини.
Прогресування захворювання характеризується певною стадийностью:
- Перша стадія – стагнація. Вона триває до 2,5 років і проявляється наступними симптомами: зниженням тонусу м’язів, їх слабкістю, уповільненням психомоторного розвитку, апатією, відсутністю інтересу до оточуючих людей і іграшок, повільним зростанням стоп і кистей в довжину, недостатнім темпом зростання голови, порушенням роботи печінки, серця, шлунково-кишкового тракту. Лікування, розпочате на такому етапі, може зупинити прогресування синдрому Ретта.
- Друга стадія – регрес триває в середньому два роки. Клініцисти діагностують цю стадію як енцефаліт. Клінічні ознаки стають більш вираженими. Дитина неспокійний, примхливий, погано спить і швидко втрачає набуті навички. На цій стадії порушується процес дихання: періодично виникає апное, з’являються ознаки гіпервентиляції – напади прискорених і глибоких дихальних рухів. Дихальні порушення виникають під час неспання хворого і зникають під час сну. Характерні неврологічні порушення: атаксія, епіпріпадкі, часті стереотипии. Цілеспрямовані руху змінюються хаотичними неконтрольованими жестами. Судомні напади супроводжуються заламування рук з різким переходом від відчуженості і беземоційності до гучного крику. Симптоматичне лікування синдрому Ретта на цій стадії залишається безрезультатним.
- Третя стадія відносної стабільності триває від 4 до 15 років. Вона відрізняється стабільним протіканням і можливим поліпшенням стану дитини. В цей час ознаки прогресування захворювання практично зникають, нормалізується емоційний фон і сон. У хворих частішають судомні напади, формується глибока розумова відсталість, з’являються гіперкінези і екстрапірамідні розлади, при яких посмикування змінюються «ступором» і онімінням.
Четверта стадія – зменшення частоти епіпріпадков аж до їх повного зникнення. Хворі втрачають рухливість, виникає атрофія м’язів, судинні порушення в ногах, трофічні виразки, різні форми викривлення хребта. Зростання тонусу м’язів в окремих кінцівках призводить до утворення контрактур. У хворих знижується маса тіла аж до крайнього ступеня виснаження. Незважаючи на істотні відхилення у фізичному розвитку, у пацієнтів відзначається повноцінне статеве дозрівання. Остання стадія спостерігається у дорослих і триває до кінця життя пацієнта.
Всі перераховані вище симптоми дозволяють визначити, на якій саме стадії знаходиться хворий з синдромом Ретта. Ознаки захворювання можуть варіюватися в залежності від швидкості прогресування хвороби і деяких індивідуальних особливостей організму.
етапи захворювання
Вперше симптоми виявляються від чотирьох місяців до 2,6 років. Але частіше від півроку до півтора років. Вчені виділили 4 стадії даної патології:
Перша стадія – стагнація. На початку хвороби стає помітним відставання в рості кінцівок і голови, з’являється млявість і знижується м’язовий тонус. Сповільнюється психомоторне розвиток, дитина втрачає інтерес до активних ігор.
Друга стадія – регрес хвороби. Погіршується стан нервово-психічного розвитку, постійний крик без заспокоєння, безсоння. Як правило, ці симптоми з’являються в період від одного року до трьох років.
Довідка! За кілька місяців вона може втратити все навички, які у нього були.
Одним з характерних симптомів захворювання вважається «миття рук», саме це нагадують руху кистями. З неврологічних розладів змінюється дихання дитини. З’являються періоди апное, які чергуються з гіпервентиляцією. Також деякі лікарі помилково ставлять діагноз аутизм, так як дитина втрачає контакт з близькими і всіма подіями, які відбуваються навколо нього. Багатьом ставлять діагноз енцефаліт, який абсолютно хибний.
Третя стадія . Цей етап захоплює вік від 3,5 до 7 років. У цей період погіршуються ті симптоми і ознаки, які з’являються на другій стадії. Ускладнюється розумова відсталість, і з’являються судоми. На перший план виходять екстрапірамідні розлади: гіперкінези, атаксія і поразки м’язів, аж до дистонії. Дитина дуже відстає від своїх однолітків за всіма параметрами. Але на тлі цього дитина емоційно стабілізується, йде на контакт і спокійно спить.
Четверта стадія – прогресування. У цей період погіршується робота рухового апарату, і прогресують порушення в русі. М’язи атрофуються, дитина стає знерухомлених, з’являється сколіоз і вазомоторні розлади. Вони відстають у рості, але статеве дозрівання при цьому залишається в межах норми. У багатьох дівчаток розвивається кахексія (тотальне виснаження організму). На цій стадії діти йдуть на контакт і емоційно стабільні, але саме в такому стані вони можуть залишатися багато років.
Дана класифікація стадій умовна. Адже не завжди вдається простежити чіткі межі між ними і часто кожен клінічний випадок унікальний. Але чітко видно прогресування хвороби без моментів поліпшення і реакції на будь-яку терапію.
етапи хвороби
- Початковий етап: від 6 до 18 місяців. На цьому етапі можуть з’явитися перші симптоми синдрому. З’являється відсутність інтересу до іграшок. Розвиток рухових навичок може бути уповільнено. Зростання окружності голови сповільнюється.
- Етап 2: від 1 до 4 років. На цьому етапі відбуваються найбільші зміни, часто швидкі, хоча і можуть бути поступовими. Дитина стає дуже дратівливим. Повторювані рухи рук стають очевидними, а цілеспрямовані руху рук губляться. Зростання окружності голови сповільнюється. Можуть виникнути проблеми з диханням.
- Етап 3: від 2 до 10 років. На цьому етапі швидка регресія другій стадії сповільнюється, і можуть спостерігатися поліпшення в поведінці, зменшується дратівливість. Контроль за рухами погіршується. Втрата м’язового тонусу ставати вираженої. Можуть початися епілептичні припадки.
- Етап 4: 10+ років. Цей етап характеризується втратою руху. Втрачається здатність ходити. Може розвинутися сколіоз. Комунікація і інтелект залишаються на колишньому рівні. Повторювані рухи рук можуть ставати менше.
Синдром Ретта у хлопчиків
В основі синдрому лежить ураження гена MECP2, який розташований в Х-хромосомі. Саме завдяки цьому гену в мозок поставляються білки і регулюється правильний розвиток. І тому, коли пошкоджується даний ген, то з’являється порушення харчування мозку, дефіцит білка і пошкоджені структури мозку просто не можуть його використовувати.
У хлопчиків одне Х-хромосома, тому вони не можуть компенсувати дане патологічний стан, як дівчатка. Ще внутрішньоутробно починаються порушення в розвитку структур мозку, тому вони вмирають практично відразу ж після народження. Захворювання не встигає розвиватися, і не виявляються симптоми, тому його важко діагностувати.
Довідка! Відомі випадки, коли народжені хлопчики з синдромом Ретта виживали.
Якщо є також синдром Клайнфельтера, при якому у хлопчика набір хромосом ХХУ. Саме завдяки другій Х-хромосомі новонародженому вдається вижити. Другим випадком, коли є ймовірність виживання, є не сильна вираженість мутації генів.
діагностика захворювання
Педіатри ставлять дитині діагноз «синдром Ретта» за клінічними проявами, які в міру дорослішання дитини стають все яскравішими. При підозрі на патологію малюка направляють на додаткові дослідження:
- КТ або МРТ головного мозку допомагає виявити вогнища ураження, які підтверджують зупинку в розвитку.
- Проводиться електроенцефалограма, яка вимірює активність мозку. Якщо є хвороба (синдром Ретта), то фіксується повільний фоновий ритм.
- УЗД внутрішніх органів показує недорозвинення нирок, печінки, серця.
- Обов’язкова молекулярно-цитологічна діагностика.
- Якщо в наявності типові прояви захворювання, призначається дослідження мутації на Х-хромосомі.
методи діагностики
Основним критерієм діагностики є збір анамнезу і з’ясування всієї клінічної картини. Але також виділяють окрему групу необхідних діагностичних критеріїв:
- адекватно протікає вагітність;
- окружність голови при народженні в межах норми, і потім відставання в рості в період з п’яти місяців і до чотирьох років;
- погіршення рухів і втрата навичок з півроку;
- мала балакучість;
- нецілеспрямовану руху рук і кистей, хлопки, посмикування, «миття рук»;
- апраксия і атаксія.
Виділяють додаткову групу:
- апное з подальшою гіпервентиляцією;
- слинотеча і аерофагія;
- судоми і атрофія м’язів;
- затримка росту і сколіоз.
Також є група виключають критеріїв:
- збільшення органів;
- ураження очей: атрофія зорових нервів і ретинопатія;
- певрологіческіе порушення;
- мікроцефалія.
Електроенцефалографія – діагностичний метод. На ньому видно повільний ритм, епілептиформні розряди.
КТ і МРТ не є інформаційними методами при синдромі Ретта. При проведенні позитронної емісійної комп’ютерної томографії видно зниження загального кровотоку в головному мозку.
діагностика
Діагностика заснована на клінічних критеріях, прийнятих в 2010 році. Розглядають цей діагноз, якщо спостерігається уповільнення зростання голови дитини. Виявити мутацію MECP2 можна за допомогою аналізу крові. Але так як такі зміни спостерігаються також при інших захворюваннях, одного дослідження недостатньо для остаточно діагнозу.
Генетичний тест може допомогти підтвердити діагноз у 80% випадків
Діферренціальная діагностика передбачає ретельні спостереження за ростом і розвитком дитини. Оскільки це захворювання зустрічається рідко, лікарі спочатку виключають такі патології, як аутизм, дитячий церебральний параліч, порушення обміну речовин і внутрішньоутробні порушення головного мозку. Генетичний тест може допомогти підтвердити діагноз у 80% випадків.
лікування
У сучасній медицині немає патогенетичного лікування синдрому Ретта, і все зводиться тільки до симптоматичного лікування для полегшення стану хворої дитини. Лікування полягає як з правильного харчування і режиму, так і з прийняття медикаментів. Наступні етапи лікування:
- Спеціальна дієта містить підвищену кількість жирів, саме завдяки їм можна підтримувати оптимальну вагу дитини, також часте годування кілька покращує і стабілізує стан.
- Спеціальна лікувальна фізкультура допоможе легше справлятися з руховими порушеннями.
- При епілептичних припадках і судомах, застосовуються антіконвульсантной терапія. Вони сприяють пригнічення вивільнення глутамату в центральній нервовій системі.
- Для корекції сну використовують мелатонін, який володіє снодійним ефектом.
- Вітаміни – сприяють поліпшенню кровообігу в головному мозку, і стимулює його роботу.
- Є спеціальні школи та заклади, де проводяться спеціалізовані програми для збереження мови і корекції рухових розладів. Особливим заспокійливим ефектом володіє музична терапія.
Важливо! Тільки кваліфікований фахівець може призначати лікування.
Вчені не перестають шукати засіб для лікування даної патології, і проводяться досліди на мишах з 15% успіхом. Можливо, незабаром лікарі винайдуть специфічне лікування.
Пропонуємо переглянути відео, яке розповідає про особливості синдрому Ретта і методиках реабілітації дітей з цим захворюванням.
Лікування захворювання: як допомогти малюкові
Специфічне лікування синдрому на сьогоднішній день ще не розроблено.
Існують обнадійливі дані про деякі лабораторних дослідженнях, під час яких вдалося «включити» поламаний ген у мишей і домогтися зникнення ознак хвороби. Практичній медицині зараз доступні лише симптоматичні методи терапії.
Медикаменти
Для полегшення стану дитини та пом’якшення симптоматики лікуючий лікар може призначити препарати наступних груп:
- ноотропні засоби для нормалізації мозкового кровообігу: Пірацетам, Церебролізин, Актовегін, Фенотропіл, Цераксон;
- антиконвульсанти для зменшення частоти і тяжкості судомних нападів: Фенобарбітал, Карбамазепін, Ламотриджин, Вальпарін;
- для корекції порушень сну: Тразодон, Мелатонін, Колиндяни, Золпідем;
- психотропні препарати: Фенибут, Гліцин, Ноофен;
- агоністи дофаміну для корекції моторних порушень: Леводопа, Бромокриптин, Толкапон, Пірибедил, Перлодел.
Крім цього, за свідченнями призначаються кошти для підтримки роботи внутрішніх органів: серця, печінки, підшлункової залози. У деяких випадках прийом протиепілептичних ліків виявляється неефективним, що дуже ускладнює лікування синдрому.
Фотогалерея – медикаменти для лікування синдрому Ретта

Пірацетам – препарат для нормалізації мозкового кровообігу

Церебролізин – ноотропний препарат для поліпшення роботи мозку
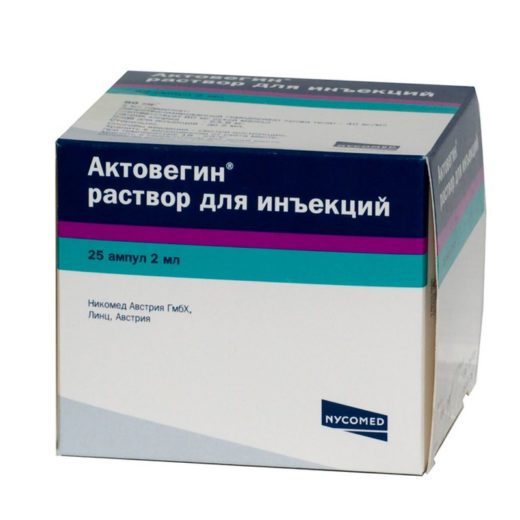
Актовегін призначають для поліпшення обмінних процесів і мозкового кровообігу
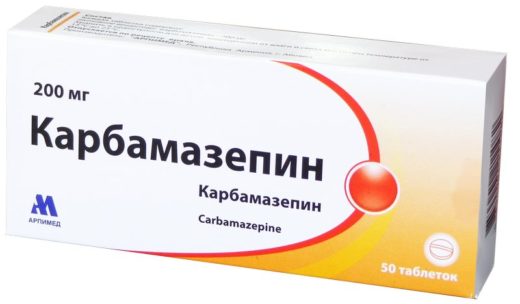
Карбамазепін належить до групи антиконвульсантов
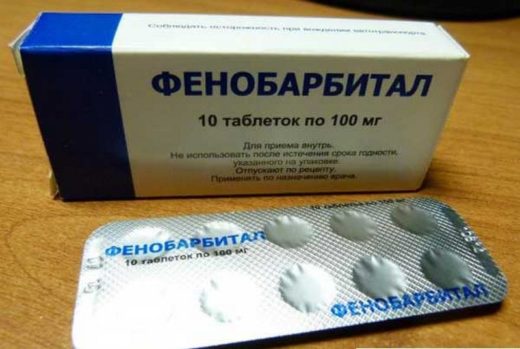
Фенобарбітал – протиепілептичний засіб з групи барбітуратів

Тразодон відноситься до групи антидепресантів і при синдромі Ретта застосовується для корекції порушень сну
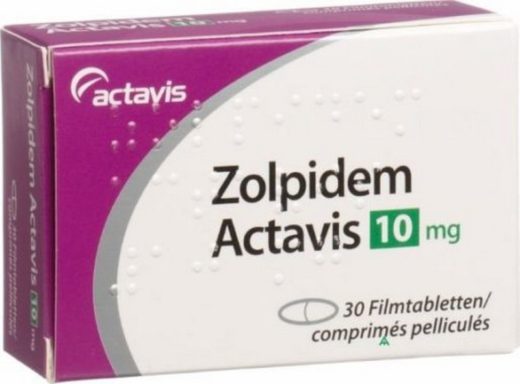
Золпідем – снодійне лікарський засіб з групи имидазопиридинов
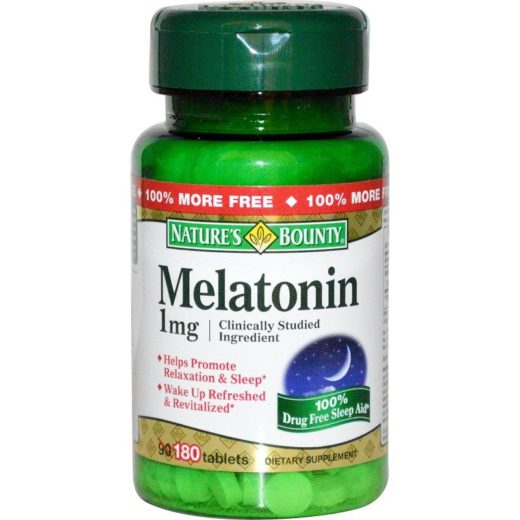
Мелатонін – основний гормон епіфізу, регулятор добових ритмів, застосовується при порушенні сну
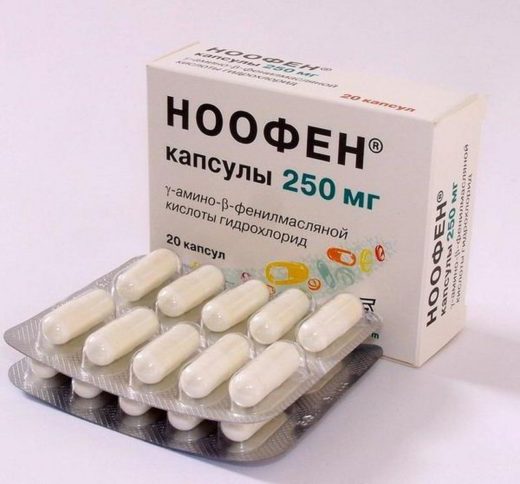
Ноофен зменшує вираженість когнітивних розладів, нормалізує сон, має властивості антиконвульсанта
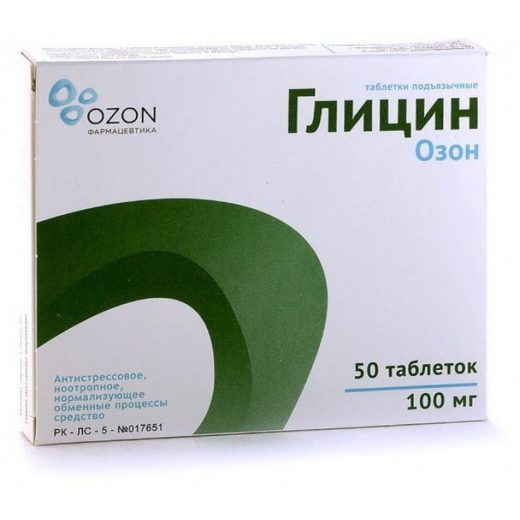
Гліцин покращує метаболізм головного мозку
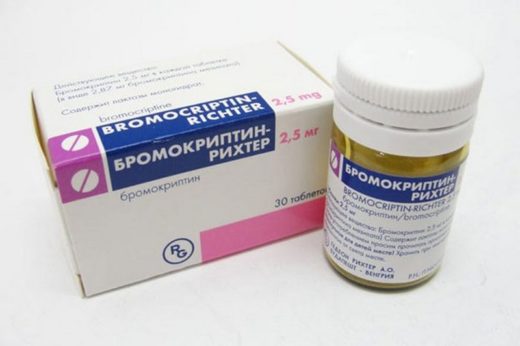
Бромокриптин застосовується при паркінсонізмі
Ускладнення і наслідки
Головним ускладненням у хворих з синдромом Ретта є нейромоторние і ортопедичні порушення, погіршення рухів, неможливість самообслуговування і пересування. Хворі втрачають здатність цілеспрямованого руху та виконання будь-яких дій.
Поява сколіозу може заподіяти біль в спині і попереку, і в цьому випадку повинен бути присутнім масаж в лікуванні.
На перших стадіях погіршується хода і в подальшому хворий зовсім перестає ходити, і прикутий до інвалідного крісла. Одним з грізних ускладнень є епілептичні припадки, які стають тільки частіше і довше за часом.
Основні симптоми захворювання
Характерним віком прояви хвороби є 7-24-місячний проміжок . Серед основних симптомів слід зазначити наведені нижче положення:
- порушення моторики верхніх кінцівок;
- порушення мови, аж до її повної втрати;
- загальмованість росту.
Медичною спільнотою досліджуване розлад включається в категорію загальних порушень розвитку. Першою характерною рисою є зупинка розвитку з подальшим погіршенням стану рухових і основних когнітивних функцій.
У міру дорослішання, відзначається виникнення таких неврологічних ознак як:
- порушення цілеспрямованих осмислених дій;
- втрата рівноваги;
- викривлення хребта.
Можливі патологічні зміни рухової активності.
На тлі розумової деградації нерідко з’являються епілептичні припадки. Поряд з цим, на відміну від схожого недуги – аутизму – досліджувані сьогодні хворі практично ніколи не виявляють схильності до самоушкодження.

Рубі-2 года.Сіндром Ретта
прогноз
Не можна точно назвати тривалість життя дітей з синдромом Ретта, кожен випадок індивідуальний. Через те, що синдром Ретта був відкритий не так давно, і методи діагностики не настільки ефективні і різноманітні, дуже важко спрогнозувати подальший стан дитини.
Деякі діти гинуть в дитинстві через ускладнення в дихальній системі, через епілепсію або дистрофії м’язів. А деякі пацієнти доживають до 30 років, що є великою рідкістю. Велика частина нерухомі і назавжди залишаються в інвалідному кріслі, але у деяких зберігаються мінімальні руху.
Важливо! При постійному догляді та симптоматичного лікування спостерігається навіть невелике поліпшення стану.
Загальні дані про порушення
Подібність цього патологічного відхилення з аутизмом видно неозброєним оком. Прості люди можуть сплутати двох хворих, один з яких -аутіст, а другий вражений синдромом Ретта. Список подібних симптомів
, За рахунок яких ці хвороби часто плутають:
- Втрата соціальних навичок.
- Емоційний або зоровий контакт з оточуючими геть відсутні.
- Занепокоєння без видимих на те причин, що супроводжується плачем і криком.
- Руки і все тіло рухаються неприродно, але однотипно.
- Кал і сеча виділяються мимоволі.
характеристика порушень
при синдромі Ретта також знаменна індивідуальними проявами, і ось їх перелік:
- Мікроцефалія.
- Кахексія.
- Патологія роботи легенів.
- Сколіоз і атаксія.
- Повна дискоординація рухів.
- Епілептичні припадки.
Немає єдиної відповіді на питання про те, скільки живуть діти, яким у спадок перейшов цю недугу. Всі випадки індивідуальні, багато в чому хід хвороби залежить від профілактики, лікування і бажання одужати.
Що потрібно запам’ятати?
- Синдром Ретта – рідкісна генетична хвороба, яка вражає Х-хромосому.
- Зміна гена MECP2 на Х-хромосомі.
- Головними симптомами є: мікроцефалія, рухові порушення, порушення координації і розумова відсталість, порушення дихання.
- Дана хвороба має 4 етапи, в яких простежується прогресування.
- Для чоловіків ця хвороба вважається фатальною і часто хлопчики при народженні вмирають.
- Основний метод діагностики – клінічна картина.
- Специфічного лікування синдрому Ретта немає, все зводиться до симптоматичної терапії.
- Найбільш грізні ускладнення: нейромоторние і ортопедичні.
- Прогноз несприятливий, через великого набору симптомів, які з роками тільки посилюються.
література
- Андрієнко Н.В. Синдром Ретта у дітей: клініка, діагностика, лікування // Автореф. дис. … канд. мед. наук. – М., 1997..
- Горбачовська Н. Л., Улас В. Ю. Всесвітній конгрес по синдрому Ретта // Журн неврол і псіхіат. – 1997. – Т. 97. – № 12. – С. 104-105.
- Грачов В.В. Синдром Ретта: питання діагностики // Журн неврол і псіхіат. – 2001. – Т. 101. – №1. – С. 22-26.
- Armstrong DD Neuropathology of Rett Syndrome // J. Child Neurol. – 2005. – V. 20 (9). – P. 747-753.
- Goyal M., O’Riordan MA, Wiznitzer M. Effect of Topiramate on Seizures and Respiratory Dysrhythmia in Rett Syndrome // J. Child Neurol. – 2004. – V. 19 (8). – P. 588-591.
- Hagberg B., Aicardi J., Dias K., Ramos O. A progressive syndrome of autism, dementia, ataxia and loss of purposeful hand use in girls: Rett’s syndrome. Report of 35 cases // Ann. Neurol. – 1983. – V. 14. – Р. 471-479.
- Percy AK, Lane JB Rett Syndrome: Model of Neurodevelopmental Disorders // J. Child Neurol. – 2005. – V. 20 (9). – P. 718-721.
Перші прояви захворювання
Якщо дитина народжується з такою патологією, то перші місяці, а іноді і роки, запідозрити її практично неможливо. При огляді педіатр не знаходить відхилень у розвитку або зростання. Іноді лікарі звертають увагу на млявість м’язів і гіпотонію, але все це списують на важкі пологи, тому не приділяють належної уваги таких симптомів. Синдром Ретта у дітей (фотографії представлені нижче) може також проявлятися наступними ознаками:
- Низькою температурою тіла.
- Пітливістю долонь і стоп.
- Блідістю шкірних покривів.
З дорослішанням малюка порушення починають виявлятися сильніше, наприклад батьки відзначають, що в віці 4-5 місяців малюк не в змозі перевернутися на спинку або пліч, а коли приходить час ходити і повзати – не може освоїти цю навичку.
особливості харчування
Харчування хворої дитини представляє певні складності. У багатьох дівчаток спостерігається підвищена слинотеча, поганий стан ротової порожнини, тому нагодувати їх справжня проблема. Деякі діти мають хороший апетит і з задоволенням вживають улюблені продукти. Але всі вони їдять дуже повільно, процес прийому їжі може розтягуватися до півтори години. Що стосується пиття, то майже у половини хворих малюків є труднощі з ковтанням, які проявляються поперхіванія, кашлем і можуть загрожувати попаданням рідини в дихальні шляхи.
- Дітям з синдромом Ретта важко пережовувати їжу, що містить грубі волокна (м’ясо, сирі овочі, фрукти), тому її потрібно подрібнювати і давати у вигляді пюре. Гарніри краще пропонувати дрібними шматочками або в Розім’яті вигляді. Під час годування потрібно стежити, щоб голова дитини була під правильним кутом, що не закидалася.
- У деяких випадках, коли процес поглинання їжі стає занадто проблематичним і болючим, має сенс годувати дитину через зонд спеціальними живильними сумішами. Такий варіант дозволяє значно поліпшити якість життя малюка і може стати справжнім для нього порятунком.
Багато дітей страждають від нудоти і часто відмовляються від їжі, тому нагодувати їх буває дуже важко, такі малюки швидко втрачають у вазі. Тому їжа повинна бути збагаченою білками і жирами, досить калорійною і вітамінізованої. Рекомендується годувати дитину малими порціями і часто (кожні 3 години), щоб не перевантажувати травну систему. Дітей молодшого віку поять вітамінізованим молоком або молочними сумішами. [Adsense3]
Класифікація
Єдине поділ такої хвороби у малюків передбачає наявність декількох етапів прогресування, в залежності від яких буде відрізнятися клінічна картина:
- 1 стадія – починається з моменту досягнення дитиною 4 місяців аж до 2-річного віку. У таких випадках починають слабо виражатися перші ознаки хвороби, чому батькам дуже важливо стежити за поведінкою дитини. Лікування, розпочате на такому етапі, може зупинити прогресування синдрому Ретта;
- 2 стадія – відрізняється прогресуванням, а клініцисти діагностують її як енцефаліт. Може маніфестувати у віковій категорії від 1 року до 3 років;
- 3 стадія – охоплює весь дошкільний період і ранній шкільний вік. Відрізняється стабільним протіканням і можливим поліпшенням стану дитини;
- 4 стадія – виявляється приблизно до 10 років. Виражається в знерухомлення дітей і формуванні вторинних кісткових деформацій.
Що це таке?
Синдром Ретта – психоневрологічне спадкове захворювання, зустрічається майже виключно у дівчаток з частотою 1: 10000 – 1: 15000, є причиною важкої розумової відсталості у дівчаток.
Вперше хвороба була описана австрійським неврологом Андреасом Реттом (нім. Andreas Rett) в 1966 році. Розвиток дитини до 6 – 18 місяців протікає нормально, але потім у дівчинки починають пропадати придбані мовні, рухові і предметно-рольові навички.
Характерним для даного стану є стереотипні, одноманітні рухи рук, їх потирання, заламування, при цьому не носять цілеспрямованого характеру. Мова не може, відповіді стають одноманітними або ехолаліческімі, часом мова зовсім пропадає (мутизм). [Adsense1]
Причини виникнення
У 90-х роках існувала гіпотеза про те, що синдром Ретта – це певний порушення, яке пов’язане з генними мутаціями, що локалізуються в Х-хромосомі; обумовлене домінантною ознакою і у хлопчиків не може поєднуватися з життям. Надалі підтвердилася передача гена-мутанта Х-хромосомою батька тим, що це спадкова патологія дуже рідко може зустрітися у хлопчиків, так як вони отримують від батька Y-хромосому. Саме тому при сімейному типі успадкування синдрому Ретта, хлопчики в таких сім’ях народжуються практично здоровими.
В даний час існують підтвердження про спадкову природу захворювання. Генетичну причину виникнення синдрому Ретта пов’язують зі зміненою Х-хромосомою і мутаціями, які відбуваються в генах, що регулюють процес реплікації. В даному випадку відбувається дефіцит деяких білків, які регулюють це зростання, а також порушується холинергическая їх функція.
Для синдрому Ретта була висунута гіпотеза про перерване розвитку, для якого характерний дефіцит нейротрофічних факторів. Таким чином, уражаються базальні ганглії, нижні моторні нейрони, втягується спинний мозок і гіпоталамус. Аналізуючи морфологічні зміни, вчені прийшли до висновку, що відбуваються уповільнення в розвитку мозку з самого народження, який повністю зупиняється в рості до чотирьох років. А також у таких дітей відзначається уповільнення в зростанні тіла і деяких соматичних органів.